
Calculating the diffusion energy barrier of a single oxygen vacancy
This is a short tutorial on the calculation of the energy barrier of diffusion for a single oxygen vacancy. The calculations are done with SOD, VTST Tools and VASP.
Introduction
Diffusion in materials plays a vital role in a wide range of applications ranging from energy storage to spintronics. This is a tutorial on generating a single oxygen vacancy and calculating the diffusion energy barrier using the nudged elastic band (NEB) method available through VTST tools along with the DFT code VASP. Before we get started here are some great articles on what I based this post on. The first article is on the Site Occupancy Disorder (SOD) method which is used to generate the vacancies.
These next ones are on the nudged elastic band method to calculate the energy barrier.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson, and G. Henkelman, “A generalized solid-state nudged elastic band method,” J. Chem. Phys. 136, 074103 (2012).
- D. Sheppard and G. Henkelman, “Paths to which the nudged elastic band converges,” J. Comp. Chem. 32, 1769-1771 (2011).
- D. Sheppard, R. Terrell, and G. Henkelman, “Optimization methods for finding minimum energy paths, J. Chem. Phys. 128, 134106 (2008).
- G. Henkelman, B.P. Uberuaga, and H. Jónsson, “A climbing image nudged elastic band method for finding saddle points and minimum energy paths,” J. Chem. Phys. 113, 9901 (2000).
- G. Henkelman and H. Jónsson, “Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points,” J. Chem. Phys. 113, 9978 (2000).
- H. Jónsson, G. Mills, K. W. Jacobsen, “Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions,” in Classical and Quantum Dynamics in Condensed Phase Simulations, Ed. B. J. Berne, G. Ciccotti and D. F. Coker (World Scientific, 1998), page 385.
Additionally, the Distortion Symmetry method can also be incorporated for generating paths taking symmetry into account. The following articles are some great resources to learn more about it.
- J.M. Munro et. al. Implementation of distortion symmetry for the nudged elastic band method with DiSPy. npj Comp. Mat. 5, 52 (2019).
- J.M. Munro et. al. Discovering minimum energy pathways via distortion symmetry groups. Phys. Rev. B. 98, 085107 (2018).
- B.K. VanLeeuwen & V. Gopalan. The antisymmetry of distortions. Nat. Commun. 6, 8818 (2015).
In the following sections, we walk through the steps required to generate the path of a single oxygen vacancy diffusion and calculate the energy required for it to occur.
Prerequisites
The following codes and scripts are used in the procedure. Please install them first. VASP needs
- SOD1
- VTST Tools2
- VASP3 - Requires re-compilation with VTST tools. Please read the VTST documentation for instructions.
- VESTA4
Other scripts used in the procedure:
Methodology
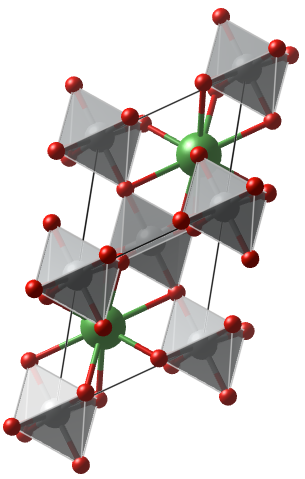
Now we follow the steps for generating the single OV and calculating the diffusion energy barrier. For this example we would be using the 10 atom R$\bar{3}c$ (spg 167) correlated nickelate LaNiO$_3$.
La Ni O
1.0
1.56361568 -2.70826180 4.30823758
1.56361568 2.70826180 4.30823758
-3.12723135 0.00000000 4.30823758
La Ni O
2 2 6
d
0.25000000 0.25000000 0.25000000 La
0.75000000 0.75000000 0.75000000 La
0.00000000 0.00000000 0.00000000 Ni
0.50000000 0.50000000 0.50000000 Ni
0.69805661 0.80194339 0.25000000 O
0.80194339 0.25000000 0.69805661 O
0.25000000 0.69805661 0.80194339 O
0.30194339 0.19805661 0.75000000 O
0.19805661 0.75000000 0.30194339 O
0.75000000 0.30194339 0.19805661 O
Generating the oxygen vacancy with SOD
-
The first step is to use the POSCAR and obtaining the WYCCAR using AFLOW. The resulting WYCCAR for this structure is given below.
La Ni O| SG: R-3c 167 PG: -3m BL: R | sym_eps: 0.019194 1.0000 5.4165 5.4165 12.9247 90.0000 90.0000 120.0000 167 2 1 1 1 Direct(WYCCAR) 0.00000000000000 0.00000000000000 0.25000000000000 La 6 a 32 -0.00000000000000 -0.00000000000000 0.00000000000000 Ni 6 b -3. 0.44805661000000 0.00000000000000 0.25000000000000 O 18 e .2 -
Next we use this in the INSOD file used to generate the vacancies using SOD. The INSOD file is displayed below.
#Title LaNiO3 single oxygen vacancy # a,b,c,alpha,beta,gamma 5.4165 5.4165 12.9247 90.0000 90.0000 120.0000 # nsp: Number of species 3 # symbol(1:nsp): Atom symbols La Ni O # natsp0(1:nsp): Number of atoms for each species in the assymetric unit 1 1 1 # coords0(1:nat0,1:3): Coordinates of each atom (one line per atom) -0.00000000000000 0.00000000000000 0.25000000000000 -0.00000000000000 0.00000000000000 0.00000000000000 0.44805661000000 0.00000000000000 0.25000000000000 # na,nb,nc (supercell definition) 1 1 1 # sptarget: Species to be substituted 3 # nsubs: Number of substitutions in the supercell 1 # newsymbol(1:2): Symbol of atom to be inserted in the selected position, # symbol to be inserted in the rest of the positions for the same species. X O # FILER, MAPPER # # FILER: 0 (no calc files generated), 1 (GULP), 2 (METADISE), 11 (VASP) # # MAPPER: 0 (no mapping, use currect structure), >0 (map to structure in MAPTO file) # # (each position in old structure is mapped to MAPPER positions in new structure) 11 0 # This section is not read if VASP files are being created # If FILER=1 then: # ishell(1:nsp) 0 core only / 1 core and shell (for the species listed in symbol(1:nsp)) 0 1 # newshell(1:2) 0 core only / 1 core and shell (for the species listed in newsymbol(1:2)) 0 0In the SOD code directory find the SGO file corresponding to the spacegroup. 167.sgo in this case. Copy it into the directory where INSOD is. Make sure it is renamed as SGO.
Then run:
sod_comb.shThis would result in a c000001.vasp file with the dummy oxygen vacancy marked with X.
vasp 1.00000000 5.416500 0.000000 0.000000 -2.708250 4.690826 0.000000 0.000000 0.000000 12.924700 La Ni X O 6 6 1 17 Direct 0.000000 0.000000 0.250000 0.000000 0.000000 0.750000 0.666670 0.333330 0.583330 0.666670 0.333330 0.083330 0.333330 0.666670 0.916670 0.333330 0.666670 0.416670 0.000000 0.000000 0.000000 0.000000 0.000000 0.500000 0.666670 0.333330 0.333330 0.666670 0.333330 0.833330 0.333330 0.666670 0.666670 0.333330 0.666670 0.166670 0.551943 0.000000 0.250000 0.000000 0.551943 0.250000 0.448057 0.448057 0.250000 0.448057 0.000000 0.750000 0.000000 0.448057 0.750000 0.551943 0.551943 0.750000 0.218613 0.333330 0.583330 0.666670 0.885273 0.583330 0.114727 0.781387 0.583330 0.114727 0.333330 0.083330 0.666670 0.781387 0.083330 0.218613 0.885273 0.083330 0.885273 0.666670 0.916670 0.333330 0.218613 0.916670 0.781387 0.114727 0.916670 0.781387 0.666670 0.416670 0.333330 0.114727 0.416670 0.885273 0.218613 0.416670 -
What we would like to do now is to migrate this oxygen vacancy to a nearby oxygen atom location as shown in the figure below. This can be done by calculating the distance between the vacancy oxygen atom (black) and a neighboring oxygen atom (red) with VESTA and selecting a nearby oxygen atom. Turn on the atom labeling in VESTA to see which atom we are considering. Update the POSCAR for the diffused structure such that this selected oxygen atom (red) is swapped with the oxygen vacancy atom (black). Let’s call the initial structure POSCAR_initial and the latter POSCAR_final.
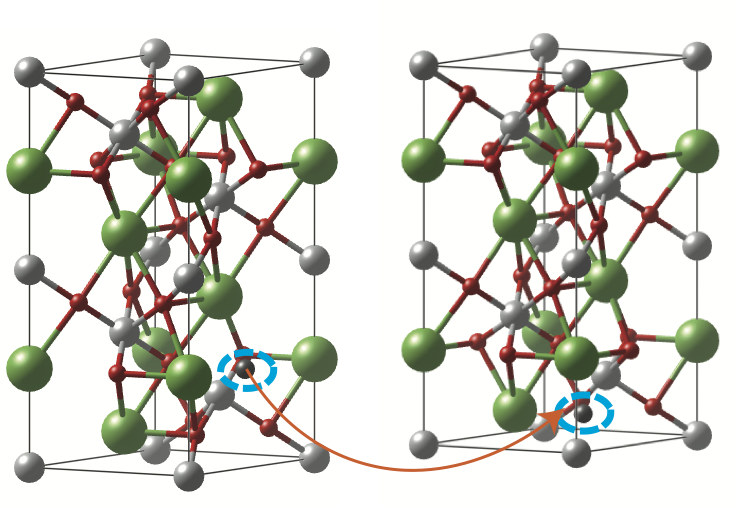
-
Now run vacancyPOSCARFormatter.py to remove the dummy oxygen vacancy and reformat the POSCAR file.
vacancyPOSCARFormatter.py POSCAR_initial POSCAR_initial_vacancyremoved vacancyPOSCARFormatter.py POSCAR_final POSCAR_final_vacancyremoved -
Optionally, phonopy6 may be used to symmetrize the structures.
phonopy --symmetry --tolerance 1E-03 <POSCAR name> -
Perform atomic relaxation on the initial and final structure. Keep the cell parameters static. I personally use my own script to call PyChemia7 to perform the relaxation.
Convergence.py relax -np 4 -incar INCARLet’s rename the relaxed structures as
POSCAR00andPOSCAR08.
Calculating the oxygen vacancy diffusion barrier with NEB
At this stage DiSPy coud be incorporated to calculate transition paths with distortion symmetry but we shall skip that for now and only perform NEB with VTST tools. VTST tools creates a linear interpolated transition path between the initial and final structure whereas DiSPy considers symmetry to generate this path(s).
-
We create a path with a certain number of intermediate images (structures) along the transition path. For this example we will create 7 intermediate images. With the two endpoints (initial and final structure) this add upto 9 total images. Run nebmake.pl in VTST scripts to generate the path.
nebmake.pl POSCAR00 POSCAR08 7 -
The atoms in the structures of the transition path may have gotten close to each other so to avoid them being too close we can set a lowerbound for the distance between the closest atoms. In our case it would be the Ni-O bond at 1.9 $\AA$.
nebavoid.pl 1.9 -
In the initial and final structure directory perform a SCF calculation to obtain the OUTCAR for each of the calculation.
-
Now in the root directory of the calculation, we would have
- 00-08 directories for the transition path each containing a POSCAR
- INCAR, KPOINTS and POTCAR files
The KPOINTS and POTCAR files are similar to the typical VASP calculation files but we now have a specialized INCAR to trigger the NEB calculation as displayed below. The important section is the NEB.
# VASP INCAR TEMPLATE # -Uthpala Herath #-------------------- STARTING PARAMETERS -------------------- system = LaNiO3 PREC = Accurate LREAL = Auto ADDGRID = .TRUE. LASPH = .TRUE. LMAXMIX = 4 # d-orbitals:4, f-orbtials:6 #-------------------- PARALLELIZATION -------------------- NCORE = 32 NSIM = 1 KPAR = 1 #-------------------- ELECTRONIC MINIMIZATION -------------------- ENCUT = 600 EDIFF = 1E-08 NELM = 100 NELMIN = 8 ALGO = Normal #-------------------- DOS -------------------- ISMEAR = 2 # Metals:2, SC/Insulators:-5, 0, Bands:0 SIGMA = 0.2 # Metals:0.2, SC/Insulators:0.05 #-------------------- SPIN -------------------- ISPIN = 1 #MAGMOM = 4*0 4*0 4*3.7 12*0 #-------------------- DFT+U -------------------- LDAU = .FALSE. LDAUTYPE = 1 LDAUL = -1 -1 2 -1 LDAUU = 0.0 0.0 2.7 0.0 LDAUJ = 0.0 0.0 1.0 0.0 LDAUPRINT = 2 #-------------------- NEB -------------------- LCLIMB = .TRUE. ICHAIN = 0 IMAGES = 7 # Excluding end points SPRING = -5.0 LNEBCELL = .FALSE. # SS-NEB Requires ISIF = 3 and IOPT = 3 # Force optimizer (Set vasp to do Damped MD with 0 time steps trigger NEB optimization) EDIFFG=-0.05 IBRION = 3 POTIM = 0 IOPT = 3 # 3:quick min, 7:FIRE NSW = 1000 ISIF = 0 LSCALAPACK = .FALSE. -
We may use a job submission script similar to the one shown below to submit the NEB calculation on a cluster. The commented sections are to perform the SCF calculation to obtain the OUTCAR for the endpoint structures unless that was done separately previously.
#!/bin/bash #---------------------------------------------------- # Sample Slurm job script # for TACC Stampede2 KNL nodes #---------------------------------------------------- #SBATCH -J OV-diffusion-vtst-relaxed # Job name #SBATCH -p long # Queue (partition) name #SBATCH -N 7 # Total # of nodes #SBATCH --tasks-per-node 32 # Total # of mpi tasks #SBATCH -t 120:00:00 # Run time (hh:mm:ss) #SBATCH --mail-type=fail # Send email at failed job #SBATCH --mail-type=end # Send email at end of job #SBATCH --mail-user=ukh0001@mix.wvu.edu ulimit -s unlimited # perform SCF calculation for end-points # cd $SLURM_SUBMIT_DIR/00 #cp ~/MatSciScripts/INCAR.scf . #time mpirun -np $SLURM_NTASKS vasp_std # cd $SLURM_SUBMIT_DIR/08 #cp ~/MatSciScripts/INCAR.scf . #time mpirun -np $SLURM_NTASKS vasp_std # run NEB calculation cd $SLURM_SUBMIT_DIR/ time mpirun -np $SLURM_NTASKS vasp_std nebresults.pl -
nebresults.pl calculates the minimum energy pathway plot (mep.eps) for the oxygen vacancy diffusion energy barrier but I wrote a Python script to suit my needs. The number of atoms for normalization can be passed in with the
-nflag.plotNEB.py -n 29The output is shown below.
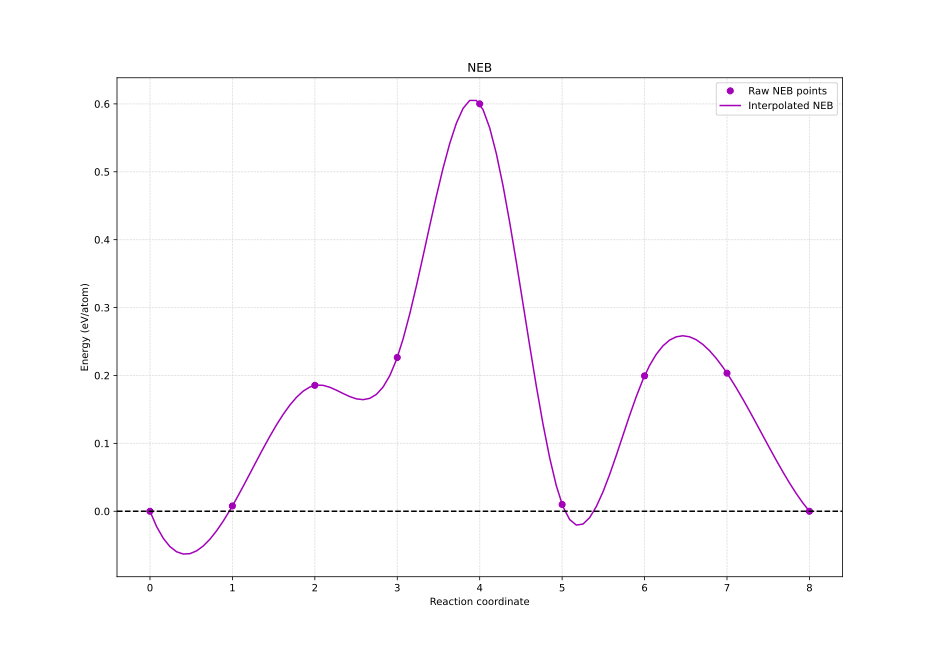
According to the plot we notice that the maximum energy required for the diffusion to occur is around 0.6 eV/atom.
-
Sometimes the calculation would not finish with the allocated time. In this case we could save the current status of the calculation and resubmit the job submission script to resume it. To save the calculation, run
vfin.pl run1where, run1 is a name for the saved checkpoint.
If you have trouble converging, play around with the force optimizer options.

Comments